
What is ANCHOR?
ANCHOR is a web-based tool whose aim is to facilitate the analysis of protein-protein interfaces with regard to its suitability for small molecule drug design.
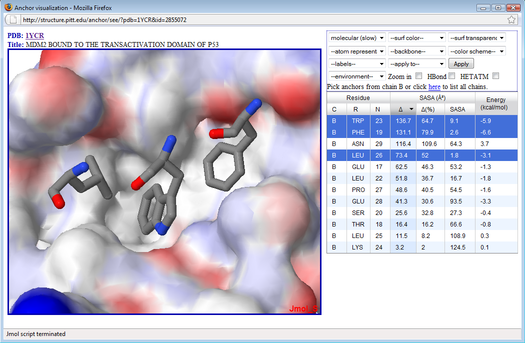
ANCHOR visualization tool
The idea behind it
Small molecule inhibitors of protein-protein interactions often bind to the hotspot areas at the protein-protein interface. The identification of hotspots residues thus constitute valuable information to be exploited on the drug design process, as they indicate the location to where small molecules have more propensity to bind. Moreover, bioisostere analogues of hotspot residues are ideal fragments to incorporate into compounds that might interfere with protein-protein interactions by mimicking the interactions encountered on the protein complex. With that in mind, ANCHOR was designed to facilitate the identification of hotspot residues at protein-protein interfaces as a starting point for designing inhibitors of protein interactions. Practitioners of fragment-based drug design may find in ANCHOR a rich and freely available source of fragments for drug discovery.
The definition of hotspot residues
Hotspot residues are usually identified experimentally as those residues that lead to a significant loss of binding affinity (ΔΔG > 1.5 kcal/mol) when mutated to alanine (ALA). Several computational methods have been developed to predict hotspot residues according to this definition. However, such definition also includes residues that are not directly involved in highly favorable interactions with the receptor protein, but, nevertheless, are required to properly stabilize the bound-like conformation of the ligand protein. We believe that these residues are not so interesting from the perspective of drug design, as they are only important on the specific context of the ligand protein. Therefore, here we (loosely) define hotspot residues as those residues burying the largest amounts of solvent accessible surface area upon binding. We call them 'anchor residues'. Intuitively, in order to achieve high binding affinities to the receptor protein, small molecules need to bury a substantial fraction of their surface area (and that is exactly what anchors do!).
How does it work?
For a given protein-protein complex submitted by the user, ANCHOR calculates the change in solvent accessible surface area (ΔSASA) upon binding for each side-chain, along with an estimate of its contribution to the binding free energy (Camacho CJ and Zhang C (2005). FastContact: rapid estimate of contact and binding free energies. Bioinformatics, 21(10):2534-6). ANCHOR does not attempt to classify the residues as 'anchor' or 'not anchor'. Instead, it lists the residues in decreasing order of ΔSASA or other criteria defined by the user such as the predicted binding energy. Then, with the assistance of a Jmol-based visualization tool, the user may interactively visualize the top ranked residues in their pockets as well as the chemical properties of the surrounding region such as hydrogen bonding.
ANCHOR database
ANCHOR includes a database of pre-computed anchor residues for about 31,000 PDB entries with at least two protein chains (but no DNA/RNA chains). For X-Ray structures, we computed the anchors from the most likely biological assembly predicted by EBI PISA. For NMR structures, we computed the anchors from the first NMR model deposited in the Protein Data Bank. The database allows query by PDB id, residue type, ΔSASA, predicted binding energy and keywords. For example, the user may query for all TRP anchor residues (residue type) contributing at least -5.0 kcal/mol (energy) and burying at least 100 A² (ΔSASA) in PDB entries related to 'cancer or oncogene' (keywords). Among the results of this query are several anticancer targets including p53-mdm2 and Smad4-Ski. Therefore, ANCHOR can also be used to quickly perform a PDB-wide search for anchor residues in protein interactions and identify protein-protein interfaces suitable for small molecule intervention.